Eine neue Analysemethode ermöglicht die automatisierte Reihenuntersuchung vielversprechender Wirkstoffkandidaten für die Arzneiforschung. Die entwickelte Technik kann die Wechselwirkung von Enzymen und anderen Proteinen mit Wirkstoffkandidaten oder anderen Molekülen rasch und in atomarem Detail zeigen. Das Konzept der „seriellen Mix-und-misch-Synchrotronkristallographie“ hat das Potenzial, das gezielte Design medizinischer Wirkstoffe auf eine neue Ebene zu heben, wie die Autoren, zu denen auch mehrere CUI-Wissenschaftler gehören, im Fachblatt „Journal of the International Union of Crystallography“ (IUCrJ) erläutern.
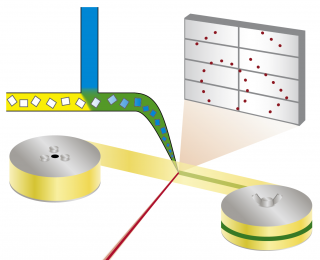
Illustration der seriellen Mix-und-misch-Synchrotronkristallographie: Wirkstoff und Proteinkristalle werden gemischt und auf ein Laufband aufgebracht, wo die einzelnen Mikrokristalle mit Röntgenlicht durchleuchtet werden. Bild: Beyerlein et al., IUCrJ [Quelle]
Zahlreiche Proteine im Körper sind lohnende Ansatzpunkte für Medikamente. Arzneimoleküle mit der passenden Form können an diese Proteine andocken und ihre Funktion gezielt abschalten oder aktivieren – je nach Zweck des Medikaments. So deaktiviert das Krebsmittel Imatinib beispielsweise gezielt eine bei bestimmten Leukämie-Formen überaktive Variante des Enzyms Tyrosinkinase, das seinerseits für die Aktivierung vieler anderer Proteine zuständig ist. Um dies zu erreichen, muss das Wirkstoffmolekül exakt in das aktive Zentrum des Enzyms passen, wie ein Schlüssel in ein Schloss. Imatinib wurde auf Grundlage der genauen räumlichen Struktur des Enzyms für diese Aufgabe maßgeschneidert.
Rationales Wirkstoffdesign
„Diese Strategie nennt sich rationales Wirkstoffdesign und ist heute eine Standardmethode in der pharmazeutischen Wirkstoffentwicklung“, erläutert der Erstautor der neuen Veröffentlichung, Kenneth Beyerlein vom Center for Free-Electron Laser Science (CFEL), einer Kooperation von DESY, Universität Hamburg und der Max-Planck-Gesellschaft. „In der Realität ist es allerdings viel komplizierter, das passende Molekül für ein Protein zu finden, als den richtigen Schlüssel für ein Schloss. Daher müssen zahlreiche potenzielle pharmazeutische Moleküle oder Teile von diesen getestet werden, was für gewöhnlich eine langwierige und komplizierte Prozedur ist.“
Um generell die Maschinerie des Lebens besser zu verstehen, interessieren sich darüber hinaus Biologen und Pharmakologen gleichermaßen für die genauen Funktionsweisen natürlicher Botenstoffe, die an Proteine andocken und damit gezielt eine Wirkung auslösen. Das von dem Team um Beyerlein und seinem DESY-Kollegen Dominik Oberthür entwickelte System bietet nun einen neuen Weg zu diesem Ziel: Es mischt Proteine in Form winziger Kristalle mit den gewünschten Molekülen, beispielsweise Wirkstoffkandidaten oder natürlichen Substraten, und macht unmittelbar darauf eine Art Röntgenbild von der räumlichen Struktur dieser Verbindung des Proteins mit dem sogenannten Liganden.
Methode der Röntgenkristallographie
Die räumliche Struktur von Proteinen erkunden Forscher meist mit der Methode der Röntgenkristallographie. Dafür muss aus dem zu untersuchenden Protein zunächst ein Kristall gezüchtet werden, der dann von allen Seiten mit Röntgenstrahlung durchleuchtet wird. Damit der empfindliche Kristall durch die intensive Bestrahlung nicht zu schnell Schaden nimmt, muss er tiefgekühlt werden. Das Röntgenlicht erzeugt charakteristische Muster, sogenannte Diffraktionsbilder, aus denen sich die innere Struktur des Kristalls und damit der räumliche Aufbau der Proteine errechnen lassen.
Um ein Protein mit einem gebundenen Liganden zu analysieren, muss jeweils entweder ein neuer Kristall aus dem Komplex gezüchtet werden, was oft ganz andere Bedingungen erfordert, oder der Kristall wird mir einer Lösung des Liganden getränkt, was nicht in jedem Fall effizient funktioniert. Selbst wenn alle Schritte in diesem Prozess mit Roboterhilfe automatisiert werden, muss für jeden Datensatz ein neuer Kristall an der Messstation montiert werden, was ein limitierender Faktor bei der Reihenuntersuchung von Protein-Ligand-Verbindungen etwa zum Aufbau umfassender Datenbanken ist.
Neue Technik nutzt Protein-Mikrokristalle
Die neue Technik folgt einem anderen Ansatz: „Wir nutzen Mikrokristalle“, berichtet Oberthür, der ebenfalls am CFEL arbeitet. „Das hat zwei Vorteile: Sie lassen sich normalerweise erheblich einfacher züchten als große Kristalle, und sie sind klein genug, damit sich ein potenzieller Wirkstoff in einer Lösung innerhalb weniger Millisekunden im ganzen Kristall verteilt und an alle darin enthaltenen Proteinmoleküle bindet.“
Das neu entwickelte System lässt einen feinen Strahl von Protein-Mikrokristallen in einer Trägerlösung auf ein dünnes Kunststoffband fließen. Dieses Tape trägt die Kristalle dann wie ein Förderband durch den Röntgenstrahl, der von einer rotierenden Blende in kurze Blitze zerhackt wird. Statt einen großen Kristall im Röntgenstrahl zu drehen und von allen Seiten zu durchleuchten, werden dabei viele Mikrokristalle in zufälliger Orientierung jeweils einmal durchleuchtet und die resultierenden Diffraktionsbilder später zu einem kompletten Datensatz kombiniert. Dieses Konzept der „seriellen Kristallographie“ ist zuerst für Untersuchungen mit Freie-Elektronen-Röntgenlasern (XFEL) entwickelt worden.
Durch eine zweite Düse in dem neuen System kann eine Lösung mit einem Wirkstoffkandidaten oder einem natürlichen Liganden zu den Kristallen fließen. Über den Punkt, an dem sich Proteinkristalle und Liganden mischen, lässt sich eine kurze zeitliche Verzögerung einbauen, etwa um einer ausreichenden Durchmischung genug Zeit zu geben oder ein bestimmtes Stadium im Bindungsprozess zwischen Ligand und Protein abzupassen.
Da dieses Verfahren keine Kühlung der Kristalle erfordert, können die Proteine bei jeder gewünschten Temperatur untersucht werden, zum Beispiel bei ihrer Arbeitstemperatur im Organismus. Auf diese Weise lässt sich auf dem ‚Mixtape‘ sogar die Bindungsdynamik zwischen Ligand und Protein verfolgen. „Wir können Chemikalien in der Sekunde mit den Proteinkristallen mischen in der wir diese mit dem Röntgenstrahl unter die Lupe nehmen und der Bindung zwischen beiden zusehen“, betont Oberthür. „Das erfordert keine neuen Wachstumsbedingungen für jeden Inhibitor und auch keinen manuellen Kristallaustausch, der ganze Prozess kann automatisiert werden.“
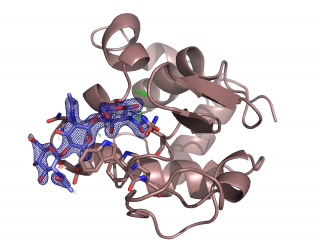
Das Enzym Lysozym (braun) mit dem inhibitierenden Zucker Chitotriose (blau). Die Untersuchung löst eine Kontroverse über den bevorzugten Bindungsort des Zuckermoleküls. Bild: DESY, Dominik Oberthür
Das Forscherteam hat sein System an DESYs hochbrillanter Röntgenlichtquelle PETRA III getestet. Für die Aufnahmen an der Messstation P11 diente das gut untersuchte Protein Lysozym als Prüfstein, zusammen mit dem Zuckermolekül Chitotriose, das die Funktion dieses Enzyms blockiert. Die hierbei verwendeten Lysozym-Kristalle hatten Durchmesser von nur sechs bis acht Mikrometern. Die mit dem neuen Aufbau gewonnenen Daten zeigen die Verbindung des Inhibitors mit dem Protein zuverlässig in hoher Detailgenauigkeit. Und obwohl die Struktur von Lysozym die erste Struktur eines Enzyms war, die auf dem Weg der Röntgenkristallographie bestimmt wurde und seit rund 50 Jahren bekannt ist, konnten die Wissenschaftler der Verbindung mit Chitotriose noch ein neues Detail entlocken: Ihre Untersuchung löst eine Kontroverse über den bevorzugten Bindungsort des Zuckermoleküls.
Erhebliche Beschleunigung
Während der erste Test des Verfahrens noch einige Zeit in Anspruch nahm, werden die wachsende Untersuchungsroutine, aber auch Fortschritte in der Detektor- und Röntgentechnik die Prozedur nach Erwartung der Wissenschaftler erheblich beschleunigen. Darüber hinaus lässt sich die Belichtungszeit für die einzelnen Mikroskristalle auf rund 100 Pikosekunden verkürzen, indem sie mit dem gesamten Spektrum des Röntgenstrahls durchleuchtet werden und nicht nur mit einer einzelnen „Farbe“, also einem schmalen Wellenlängenbereich, wie es zurzeit üblich ist. Das hat eine andere Studie kürzlich gezeigt. Eine Pikosekunde ist eine billionstel Sekunde.
„Wir entwickeln Methoden, um die Struktur gebundener Proteine für die Medikamentenforschung mit hohem Durchsatz zu erkunden“, fasst Beyerlein zusammen. Da Messzeit an den vergleichsweise zahlreichen Synchrotron-Strahlungsquellen in deutlich größerem Umfang verfügbar ist als an den wenigen, relativ neuen Röntgenlasern auf der Welt, sehen die Wissenschaftler in ihrem Verfahren das Potenzial, für die Medikamentenentwicklung routinemäßig große Bibliotheken potenzieller Inhibitoren und Wirkstofffragmente zu durchsuchen. „Dies automatisiert und damit deutlich schneller als mit konventionellen Methoden zu tun, wäre ein großer Schritt vorwärts für das strukturbasierte Wirkstoffdesign“, betont Beyerlein.
An der Arbeit waren Wissenschaftler der California State University, der Norwegischen Hochschule für Wissenschaft und Technologie, der Universität Uppsala, von Europas neuem Röntgenlaser European XFEL, vom Exzellenzcluster „The Hamburg Centre for Ultrafast Imaging“ (CUI), der Universität Hamburg, der Universität Oxford, vom US-Beschleunigerzentrum SLAC, der Fachhochschule Lübeck und von DESY beteiligt. Text: DESY, Red.
Originalarbeit:
Kenneth R. Beyerlein, Dennis Dierksmeyer, Valerio Mariani, Manuela Kuhn, Iosifina Sarrou, Angelica Ottaviano, Salah Awel, Juraj Knoska, Silje Fuglerud, Olof Jönsson, Stephan Stern, Max Wiedorn, Oleksandr Yefanov, Luigi Adriano, Richard Bean, Anja Burkhardt, Pontus Fischer, Michael Heymann, Daniel A. Horke, Katharina E. J. Jungnickel, Elena Kovaleva, Olga Lorbeer, Markus Metz, Jan Meyer, Andrew Morgan, Kanupriya Pande, Saravanan Panneerselvam, Carolin Seuring, Aleksandra Tolstikova Julia Lieske, Steve Aplin, Manfred Roessle, Thomas A. White, Henry N. Chapman, Alke Meents, and Dominik Oberthuer
“Mix-and-diffuse serial synchrotron crystallography”
IUCrJ, 2017
DOI: 10.1107/S2052252517013124